|
Pulmoner Vasküler ve Erişkin Doğumsal Kalp Hastalıkları Çalışma Grubu
Yönetim Kurulu
Başkan:
Prof. Dr. Bahri Akdeniz
Üyeler
Halil Ataş
Mehmet Kaplan
Ergün Barış Kaya
Murat Meriç
Gülten Aydoğdu Taçoy
Katkıda Bulunanlar:
Dr. Ayşe Çolak
Dr. Özden Seçkin Göbüt
Dr. Ümit Sinan Yaşar
|
|
|
 
PH Gündem - Gebeliğin ortaya çıkardığı Pulmoner Hipertansiyon (Dr. Özden Şeçkin Göbüt, Dr. Gülten Aydoğdu Taçoy)Gebeliğin ortaya çıkardığı Pulmoner Hipertansiyon
Dr. Özden Şeçkin Göbüt, Dr. Gülten Aydoğdu Taçoy OLGU 2
Gebeliğin ortaya çıkardığı Pulmoner Hipertansiyon
Dr. Özden Şeçkin Göbüt, Dr. Gülten Aydoğdu Taçoy
Gazi Üniversitesi Tıp Fakültesi, Kardiyoloji Anabilim Dalı
Olgu Sunumu
26 yaşında kadın hasta eforla ortaya çıkan çarpıntı ve nefes darlığı yakınmaları ile başvurdu. Hastanın öyküsünden 2 ay önce C/S ile doğum yaptığı ve şikayetlerinin doğum sonrasındaki ilk günde başladığı öğrenildi. Hastanın çocukluk döneminden beri çabuk yorulma yakınmasının da olduğu öğrenildi. Hastanın doğum sonrası şikayetleri başlayınca başvurduğu dış merkezde tetkikleri yapılmış ve transtorasik ekokardiyografisinde sağ kalp boşlukları geniş ve maksimum sistolik pulmoner arter basıncı s(PAB) 49 mmHg saptanarak ileri tetkik için merkezimize gönderilmiş. Hastanın özgeçmişinde Guatr nedeniyle Tiroid hormon replasman tedavisi aldığı belirlendi. Hastanın fizik muayenesinde patolojik bulgu olarak boyun ven dolgunluğu, kalp muayenesinde palpasyonla parasternal lift, kalp sesleri takikardik, sternum solunda 2/6 sistolik üfürüm saptandı. Hastanın istirahat oksijen satürasyonu %97 ölçüldü. Hastanın elektrokardiyografisinde NSR (Şekil 1), sağ ventrikül hakimiyetini düşündüren V1-3’te T negatifliği saptandı. Laboratuvar tetkiklerinde tam kan sayımı ve biyokimyasal parametreleri normal saptandı. BNP 1500 pg/mL saptandı. PA Ac grafisinde kardiyomegali, pulmoner konus dilatasyonu saptandı (Şekil 2). Transtorasik ekokardiyografide sağ kalp boşlukları geniş, sPAB 87 mmHg, sağ ventrikül S velosites, 8 cm/s, TAPSE 13 mm, RV/LV>1, IVC 16 mm saptandı. Hastada PHT tanısı açısından Romatolojik belirteçler değerlendirildi ve N saptandı. Hastanın Solunum Fonksiyon Testi ve Difüzyon Kapasitesi değerlendirmesi neticesinde Grup III dışlandı. Hastada mevcut bulgularla Grup II PHT olmadığı belirlendi. Hasta PHT etiyolojisi açısından serviste yatırılarak izlenirken, 3 dakika kadar süren epileptik atak geçirdi. Nörolojik olarak değerlendirilen hastanın Beyin BT Normal bulundu. EEG’de patoloji saptanmadı. Hastanın senkop yakınmasının PHT ile ilişkili olduğuna karar verildi. Hastanın D Dimer düzeyi normal olmakla birlikte dış merkezde yapılmış Ventilasyon Perfüzyon sintigrafisi PTE ile uyumlu gelmişti, kontrol VPS’de ise tek bir segmentte mismatch izlendi. Bunun üzerine hastanın tedavisine antikoagülan tedavi eklenemesine karar verildi. Fakat hastadaki tek segment tutulumu ile Pulmoner HT ciddiyetinin uyumsuz olması nedeniyle etiyolojiye yönelik tetkiklere devam edildi. Hastaya yapılan sağ kalp kateterizasyonunda PA 87/33/51 PKKB 7 PVR 6,8 WÜ ölçüldü. Hastaya yapılan vazoreaktivite testinde İnhaler İloprost ile PAB 33/13/20 PVR 2,3 WÜ’e düşmesi üzerine hastada Vazoreaktif PAH saptandı. Hastaya Nifedipin 2x30 mg ardından 2x60 mg dozuna geçildi. Birinci ay kontrolünde FK Sınıf I’e kadar düzelme gösterdiği ve BNP değerinin ise 74 olduğu saptandı. TTE de sPAB 40 mmHg saptandı, diastolik disfonksiyonun düzeldiği saptandı. Hastanın durumun stabilitesi nedeniyle kontrol sağ kalp kateterizasyonunun 6. ayda yapılmasına karar verildi. Kontrol perfüzyon sintigrafisinde mismatch saptanmadı.
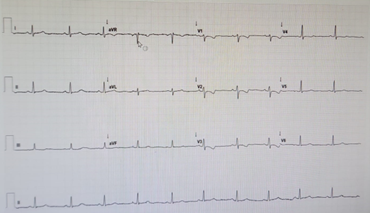
Şekil 1 - Elektrokardiyografi
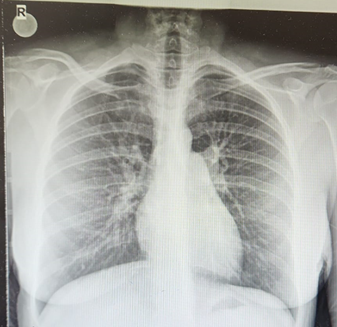
Şekil 2 - PA Ac grafisi
Tartışma
Pulmoner hipertansiyon; normal kalp debisinin varlığında, ortalama pulmoner arter basıncının (PAB) 20 mmHg üzerinde olmasıdır. Pulmoner arteriyel hipertansiyon (PAH) ise, klinik pulmoner hipertansiyon sınıflandırılmasında Grup 1’de yer alan ve pulmoner arteriyollerde proliferasyon, fibrozis, vazokonstriksiyonu ve remodelling ile karakterizedir.1-3 Pulmoner vasküler hastalık sonucunda pulmoner vasküler direnç yükselir. Sonuçta ciddi sağ kalp yetmezliği ve ölüme yol açar 4 PAH terimi; idyopatik formların yanı sıra, yine ilerleyen süreçte aynı histopatolojik değişikliklere yol açtığı gösterilmiş; kalıtsal pulmoner hipertansiyonu, ilaç ve toksinlere maruziyeti, HIV enfeksiyonuna maruziyeti, yenidoğan inatçı pulmoner hipertansiyonu, konjenital kardiyak anomalileri, kollajen vasküler hastalıkları ve portal hipertansiyonu kapsamaktadır.1
İPAH hastalarında klinik seyir oldukça değişkendir. Yapılan kapsamlı çalışmalar ortalama sağ atriyal basınç ile ortalama pulmoner arteriyel basıncın mortalite ile güçlü ilişkisi olduğunu göstermiştir. Ek olarak NYHA fonksiyonel sınıflaması da mortalite için güçlü bir prediktiftir. Kapsamlı veriler mortalitenin en sık sağ kalp yetmezliği ile ilişkili olduğunu göstermektedir.4 Ayrıca hastalarda enfeksiyon, kanama gibi takipte gelişebilen komplikasyonlar gözlenebilir.5 Kötü sağkalım oranları göze alındığında hastalığın doğru yönetimi oldukça kritiktir. İyi alınmış bir anamnez ve ayrıntılı bir fizik muayenenin ardından elde edilen göğüs radyogramı ve transtorasik ekokardiyografi bulguları, hastalık için süphe uyandırmada değerli olsa dahi; kesin tanı ve doğru takip için bazal sağ kalp kataterizasyonu (SKK) ile invaziv hemodinamik değerlendirme şarttır. SKK ile tanıyı kesinleştirme ve diğer nedenlerin ekartasyonunun yanı sıra; hastalığın ciddiyeti ve prognozu da belirlenmiş olur.6,7
İdyopatik, kalıtsal veya ilaç ilişkili PAH tanısı olduğu varsayılan hastalarda SKK ile eş zamanlı olarak vazoreaktivitenin de mutlaka test edilmesi gereklidir.8,9 Bu sayede kalsiyum kanal blokörlerine (KKB) olumlu uzun dönem yanıt gösterebilecek vazoreaktif alt küme belirlenir. Vazoreativite testi için çeşitli vazodilatatörler kullanılabilir. Vazodilatör ajan olarak İV Epoprostenol, inhale NO veya iloprost önerilir. NO yüksek dozda kardiyak debiye etkisi düşük olan hızlı etkili bir ajandır. Ancak önemli bir dezavantajı bazı hastalarda rebound şekilde pulmoner hipertansiyonu arttırmasıdır. İloprost ise kalp debisine çok az etki eder, seçici olarak pulmoner arter basıncını etkiler ve kronik tedavi olarak da endikedir. Dezavantajı en az olan vazodilatatördür. İntravenöz epoprostenol ise inhale NO veya inhale ilioprost yokluğunda bir alternatiftir. İntravenöz epoprostenol aynı zamanda kronik tedavide de yeri olan bir ajandır ancak en önemli dezavantajı sistemik ciddi hipotansiyon ve ayrıca vazoreaktivite testinde kullanımında belli aralıklarla doz arttırılması gerekli olacağı için işlemin uzun sürmesine neden olmasıdır.10,11 Vazoreaktivite testinin pozitif olması, artmış veya normal kalp debisi ve etkilenmemiş kalp hızı varlığında, ortalama pulmoner arter basıncında >%10 mmHg azalma ve beraberinde ortalama PAB mutlak değerinin <40 mmHg olmasıdır.10,11 Vazoreaktif saptanan hastalarda ilk seçilecek tedavi kalsiyum kanal blokörleri olmalıdır. PAH'ta ağırlıklı olarak kullanılan KKB'ler felodipin, nifedipin, diltiazem ve amlodipindir. Ancak yine de vazoreaktivite gösteren bazı hastalar KKB’lerine iyi yanıt veremeyebilir veya sadece kısıtlı bir süre yanıt verebilir. Bu nedenle hastaların yakın takibinin önemi de unutulmamalıdır. Daha da önemlisi tam fayda için KKB’lerin titre edilerek önerilen dozlarda kullanılmasıdır. Örneğin amlodipin 5 mg ile başlangıç sonrası 15-30 mg/gün, nifedipin 30 mg başlangıç sonrası 60-180 mg/gün, diltiazem 120 mg başlangıç sonrası 120-360 mg/gün.11 Vazoreaktivite gösteren ve KKB ile önerilen dozda tedavi edilen hastalara, 3-6. ayda yeniden değerlendirme önerilmektedir. Klinik olarak fonksiyonel kapasitesi iyileşen ve hemodinamik olarak ortalama PAB’ı <30 mmHg, PVR <4 WU sağlanan hastaların kronik yanıtı yeterli olarak kabul edilir. Tatmin edici bir yanıt alınamadığı takdirde ek PAH tedavisine başlanmalıdır. Ancak bu durumda unutulmaması gereken her ne kadar KKB’ne yanıt tatminkar olmasa da; KKB tedavisini sonlandırmak, klinik kötüleşmeye neden olabilir. KKB'lerin onaylı PAH ilaçlarıyla kombinasyonu bazı hastalarda gerekli olabilir. Vazoreaktivite çalışması yapılmamış veya testi negatif olan hastalarda, diğer endikasyonlar için standart dozlar reçete edilmedikçe, ciddi yan etkiler (şiddetli hipotansiyon, senkop ve RV yetmezliği) potansiyeli nedeniyle KKB'lere başlanmamalıdır.11
Sunduğumuz vazoreaktif PAH olgumuzda, nifedipin dozu hastamızın tolere edebileceği yüksek doza çıkarıldı ve erken dönemde klinik olarak dramatik olumlu yanıtlar alındı. NYHA fonksiyonel kapasitesinde ve ekokardiyografi değerlendirmesinde belirgin düzelme izlenen hastamıza 6. ayda kontrol SKK planlandı. IPAH hastalarında ilk SKK sırasında yapılan vazoreaktivite testlerinden, %10-20’sinin pozitif olduğu görülmüştür. Ek olarak uygun yüksek dozda verilen KKB ile ortalama pulmoner arter basıncında belirgin düşüş olacağı, dahası bu durumun 20 yıldan fazla sürebileceği de bildirilmiştir. Bu husus İPAH hastalarında; belirli bir alt kümede, pulmoner hipertansiyonun geri döndürülebilme olanağının olduğunu ve yaşam kalitesinin yanı sıra yaşam süresinin de artabileceğini kanıtlamaktadır.5,12 Daha karmaşık olan ve netleşmemiş bir husus ise İPAH’da vazoreativitenin pozitif veya negatif olduğu farklı alt grupların olup olmadığı veya bu yanıtın aynı hastalığın farklı evreleri olup olmadığıdır. Ancak vazoreaktivite testi negatif olan hastalarda verilen KKB tedavilerinin kısa veya uzun dönemde hiçbir fayda sağlamıyor gibi gözükmesi bu antitenin içten içe farklı hastalık alt gruplarına özgü olduğunu desteklemektedir.5
Kaynaklar
- Galiè, Nazzareno, et al. "2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT)." European heart journal 37.1 (2016): 67-119.
- Blaise, Gilbert, David Langleben, and Bernard Hubert. "Pulmonary arterial hypertension: pathophysiology and anesthetic approach." The Journal of the American Society of Anesthesiologists 99.6 (2003): 1415-1432.
- Chan, Stephen Y., and Joseph Loscalzo. "Pathogenic mechanisms of pulmonary arterial hypertension." Journal of molecular and cellular cardiology 44.1 (2008): 14-30.
- Kylhammar, David, et al. "A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension." European heart journal 39.47 (2018): 4175-4181.
- Bonow, Robert O., et al. Braunwald's heart disease e-book: A textbook of cardiovascular medicine. Elsevier Health Sciences, 2011.
- Simonneau, Gerald, et al. "Clinical classification of pulmonary hypertension." Journal of the American College of Cardiology 43.12S (2004): S5-S12.
- Hoeper, Marius M., et al. "Definitions and diagnosis of pulmonary hypertension." Journal of the American College of Cardiology 62.25S (2013): D42-D50.
- Barst, Robyn J., et al. "Diagnosis and differential assessment of pulmonary arterial hypertension." Journal of the American College of Cardiology 43.12S (2004): S40-S47.
- Ishii, Satoshi, et al. "Prognostic value of follow-up vasoreactivity test in pulmonary arterial hypertension." Journal of Cardiology 82.1 (2023): 69-75.
- Malhotra, Rajeev, et al. "Vasoreactivity to inhaled nitric oxide with oxygen predicts long-term survival in pulmonary arterial hypertension." Pulmonary circulation 1.2 (2011): 250-258.
- Humbert, Marc, et al. "2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: Developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG)." European heart journal 43.38 (2022): 3618-3731.
- Galie, Nazzareno, et al. "Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT)." European heart journal 30.20 (2009): 2493-2537.
|
